
Battling the CF Monster
By Laura Stephenson Carter
Photographs: Jon Gilbert Fox (email)
Physiologist Bruce Stanton leads one of the country's major research groups attacking the ravages of cystic fibrosis. Dozens of scientists, clinicians, and outcomes experts are honing in on new treatments, new medications, and a new understanding of the genetic disease.

|
|
Bruce Stanton and, in the inset, some of the DMS troops who are waging battle against CF |
It's so neat to go to cocktail parties and say you manipulate bacteria with lasers," says second-year graduate student Niles Donegan. Donegan, who is studying a bacterium that colonizes the lungs of patients with cystic fibrosis (CF), has been enthusiastically describing his research.
"Oh Niles," sighs his lab-mate and fellow graduate student Todd Jarry, giving a mock-exasperated roll of his eyes.
"I know—geeks," admits Donegan with a laugh.
Yet it's "geeks" like Donegan and Jarry who, over the past two decades, have extended the life expectancy for CF patients from the mid-teens well into adulthood. The CF research group at Dartmouth —one of the country's major integrated CF research collaboratives —is exploring the inner workings of CF cells and their bacterial invaders and seeking better medications and other therapies for CF. The researchers' hope is that maybe, someday, there will be a way to prevent or cure the devastating genetic disease.
CF is a chronic, progressive disorder caused by inheriting two copies of a defective gene. As a result, the body's mucous glands—in the respiratory, digestive, and reproductive systems—and sweat glands don't work properly. People with the disease are prone to chronic lung infections and are unable to absorb fats and other nutrients from food. Before effective antibiotics were available, most people with CF died in early childhood, but today many survive well into their adult years. In the U.S., about 30,000 children and adults have CF and more than 10 million people carry one copy of the defective CF gene.
Integration
At DMS, Professor of Physiology Bruce Stanton, Ph.D., facilitates an impressive group of CF researchers and clinicians. Dartmouth has "a comprehensive research program that includes basic science research, clinical research, patient care and education, and training for M.D. and Ph.D. fellows," says Stanton. "We're an integrated group."
That integration benefits both physicians and researchers. "We have a clinical expertise, they have a bench-research expertise," says pulmonologist Worth Parker, M.D., who directs the adult program at DHMC's CF Center, while pediatrician Bill Boyle, M.D., directs the center itself. "We try to cross-pollinate," adds Parker. "We provide feedback to them about the clinical aspects of CF, which they don't know as much about." For instance, a researcher might ask Parker whether CF patients would comply if asked to take a nebulized medication more than twice a day. "And I'll say, 'No, keep it twice a day or less and they may do it,'" Parker says.
Microbiologist George O'Toole, Ph.D., describes another plus of the Dartmouth group's collegiality: "If we need samples from patients to do certain experiments, there's the ability to do that. . . . It's probably not the biggest CF group in the country, but it's a very interactive group. All of us work together very well."
Stanton began doing basic CF research soon after his 1984 arrival at DMS. Now, the Dartmouth group is funded by many organizations, including the Cystic Fibrosis Foundation. The latter group, says Stanton, has underwritten "a cell biology core" for the program.
"The CF community counts on [Stanton] to move the science forward in his own lab, but his contributions do not stop there," says Preston Campbell, M.D., executive vice president of the CF Foundation. "He is also an excellent leader and effectively brings other investigators together to work as a team to conquer this disease."
Living with cystic fibrosis
Jonathan Dailey is 25 years old, lives with his parents in Nashua, N.H., and is working toward a bachelor of fine arts degree at the New Hampshire Institute of Art in Manchester. He hopes to someday work in advertising design.
When he was five, he was diagnosed with cystic fibrosis. Since 1992, Dailey has been under the care of DHMC's Worth Parker or of physicians at the CF Clinic in Manchester.
When he was little, however, "doctors could never pinpoint what was wrong with me," says Dailey. "My parents were always told I had allergies, bronchitis, or pneumonia." It wasn't until he had a rectal prolapse that ER doctors suggested to his parents that he be tested for CF. His parents took him to Children's Hospital in Boston and "within a half-hour of the sweat test, they told my parents that I had CF," Dailey says.
Although doctors told him he wasn't likely to live through high school, medical treatments for CF have improved significantly over the past 20 years. He not only made it through high school but graduated 35th in a class of 273, even though his illness caused him to miss a lot of school.
To stay relatively healthy, Dailey has to take a number of medications each day. Some are antibiotics to fight the constant infection in his lungs, some help to thin the mucus in his lungs, some help him digest his food, and others help him cope with other symptoms of the disease. The list includes Pancrease MT16, Dicloxacillin, Bactrim, calcium supplements, vitamins C and D, an ADEK multivitamin, Colchicine for joint discomfort, albuterol nebulizer treatments, Pulmozyme nebulizer treatments, TOBI nebulizer treatments, an MDI inhaler when he needs it, and oxygen at night to help him sleep.
"For the most part, I've been on them for so long that I don't even remember if there were side effects when I first started taking them," Dailey says. "My body's so used to taking them now that it's just natural."
In addition to taking his drugs and vitamins, Dailey does three to five breathing treatments a day, followed by autogenic drainage, an exercise he was taught at the DHMC CF Clinic. "It's where you take in different amounts of air and you force it out, making 'H' sounds, huffing sounds. It helps loosen the mucus so [you] can cough it out a lot easier."
Dailey also tries to exercise when he can, but he can't run and he tires easily just going up and down stairs. "Exercise is important, but I'm a real slacker in that department," he admits. He works out some with hand weights and has a Cardioglide exerciser, too.

|
|
The shadow of cystic fibrosis is ever-present for Jonathan Dailey, but with the help of more than a dozen medications (some of which are pictured below) he is able to lead a near-normal life. He's working toward a fine arts degree and also enjoys baby-sitting regularly for his fiancee's sixyear- old daughter, Savannah (pictured at right). |
"Over the past 10 years I have really noticed a slight decline in my health," Dailey says. "I've gone in the hospital more frequently, I cough a lot more . I have more sputum production on a regular basis . I have noticed a decline in lung function. I can't do the things I used to do — run around, laugh without coughing, walk up stairs without resting . . . I have more aches and pains now. I am taking more meds than I used to.

|
"I see others my age working or going to school full-time," he says, "and I have a hard time keeping up with one or two classes."
Dailey is not a complainer; he's just stating facts. He finds that coughing "is the worst part of CF, because you are constantly doing it and you cannot control it, no matter how much you try. I have a hard time being around new people, strangers, or in certain places [when] I have to cough. People think that you have a cold or you smoke. Try and sit through a movie at the movie theater and have to cough. It usually is loud and sounds really bad. Sometimes I go through coughing fits where I cough and cough and cough . . . usually followed by pressure headaches."
He's been hospitalized so many times for CF that he's lost count. The first time was for two weeks when he was eight years old. He's usually "hospitalized every two to three months for IV cleanouts," he says, but adds with pride that he hasn't had to go in for the past 14 months. And although he'd been considering a lung transplant, he has felt well enough recently to take himself off the waiting list for now.

|
All in all, Dailey remains optimistic about his future. "I have found the woman of my dreams," he concludes, noting that he and his fiancee, Melissa Amick, plan to be married in April of 2001.
Photographs of Jonathan Dailey by Mark Austin-Washburn.
Mucus and mutations
Some DMS researchers are looking into defects in ion trafficking within CF cells, while others are investigating the bacteria that colonize the lungs of CF patients.
In people with CF, an abnormality in lung and pancreas cells creates a thick, sticky mucus that clogs the bronchial tubes in the lungs and blocks the ducts and tubes leading from the pancreas to the intestine. In 1989, the culprit was identified—a mutated gene. When the gene is working properly, it produces a protein responsible for the movement of salt (sodium and chloride) across cell membranes. In lung cells, salt moves through chloride channels in the cell membrane to the airway surface. Then, just as a dry sponge soaks up water, the chloride and sodium combination pulls water out of the cells to create a thin fluid layer that coats the airway surface and keeps the cilia moist so they can do their job of moving foreign particles (such as bacteria) along the airway and out of the lungs. But in the lungs of a CF patient, the salt and water balance is out of whack: mucus coats the bronchioles, making it nearly impossible for the cilia to clear bacteria from the airway.
|
In people with CF, an abnormality in lung and pancreas cells creates a thick, sticky mucus that clogs the body's ducts and tubes. In 1989, the culprit was identified—a mutated gene. |
Intriguingly, the CF mutation may not be all bad. Some scientists believe there may be a selective advantage to having just one copy of the defective gene, since it may protect against the often fatal diarrhea of cholera. People with one abnormal CF gene seem to have half as many secretory chloride channels in their intestine, so they would suffer only half as much secretory diarrhea.
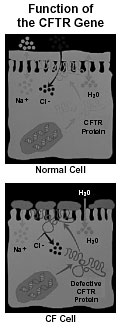
"CF is caused by a mutation in a single gene— the CFTR gene," explains Stanton. CFTR stands for cystic fibrosis transmembrane conductance regulator. There are more than 800 ways the CF gene can be mutated. Some mutations prevent the CFTR protein from being made, others keep it from regulating chloride transport, and the most common one—which scientists call DF508 (delta F508)—causes the protein to misfold so it can't get to the cell membrane.
"We have a really strong group here that works on . . . DF508," says Stanton. He explains that "delta" means deletion. "It's a deletion of F, meaning phenylalanine, [and] 508 means it's the 508th amino acid. . . . There are 1,480 amino acids [in CFTR]," he adds. The misfolding caused by this mutation means that the protein, which works only if it can get to the cell membrane, remains stuck in the cell.
Fundamental error
Stanton says the DF508 research group—which includes his own laboratory, plus "George Langford in biology and Joshua Hamilton in pharmacology—[is] very interested in trying to understand the basic cell biology of how DF508-CFTR folds normally and how it moves through what is called the biosynthetic pathway.
|
|
The cystic fibrosis transmembrane conductance regulator (CFTR) protein regulates channels that allow the negatively charged chloride (Cl) ions to exit airway cells. Normally, the positively charged sodium (Na) ions follow the Cl, and the sodium-chloride combination pulls out water to line the airway with fluid. But in CF cells, the defective CFTR protein blocks the Cl channels, so too much sodium and water are pulled into the cell, leaving behind dry mucus. |
"There are probably a dozen other diseases that have the same basic, fundamental error," Stanton continues. Alzheimer's, Tay-Sachs, Creutzfeldt-Jakob (the human variant of "mad cow" disease), and other prion diseases are a few of the conditions that involve a misfolded protein. "Anything we find and understand about how the CFTR protein doesn't fold properly is probably going to have implications for all these other diseases as well."
Before pharmacologist Joshua Hamilton, Ph.D., got involved with CF, he was a cancer researcher trying to figure out how some cancer cells were able to develop resistance to chemotherapy agents. His lab discovered that certain drugs could turn off a gene that codes for drug resistance—p-glycoprotein, which happens to be a cousin of CFTR.
Stanton suggested that Hamilton see whether he could get similar results with CFTR. The next thing Hamilton knew, "We suddenly found ourselves doing CF research."
Two chemotherapy agents were tested—mytomycin C and doxorubicin (known as "dox")—says Rangan Maitra, a graduate student in Hamilton's lab. "With mytomycin C we didn't see a very large effect. But . . . if you treat cells with doxorubicin, you can see this functional enhancement of chloride secretion." And it takes a lot less dox to correct the CFTR problem than it takes to turn off the gene that makes cancer cells resistant to chemotherapy agents.
"What really, really excites me at this point is trying to figure out how these drugs are working," says Maitra. "We know that it promotes DF508 expression at the plasma membrane. . . . How it really happens at the molecular level is yet to be characterized."
Hamilton suspects dox won't prove to be a cure for CF. But, he says, "we'd look for a chemical that's closely related. We hope to find something as effective as dox, without the toxicity problem."
Excess sodium
The CF mutations are responsible for still more physiological problems. "The story is more complicated than just CFTR," says Stanton. "Not only do you lose chloride and water secretion, but you get an excess sodium reabsorption, and that causes the airways to become even more dehydrated."
This hyperabsorption of sodium is the province of pharmacologist Frank Gesek, Ph.D. He's looking at how elevated levels of a cytokine called Tumor Necrosis Factor (TNF) cause airway epithelial cells to absorb too much sodium. This particular cytokine is normally secreted by lymphocytes and macrophages, as an immune-system response to invading bacteria, but Gesek found that airway epithelial cells release TNF, too. The TNF enhances sodium transport, which further dehydrates the lungs and makes them more susceptible to infection, so, in turn, the cells produce more TNF, "and it keeps going further and further until you just basically run down your lung function," Gesek says.
His lab is trying to figure out how the TNF-activated receptor in the cell signals for the sodium transport gate to open. "It's the first time we've been able to link a cytokine that's been demonstrated in a number of different diseases—cystic fibrosis, diabetes, arthritis, Crohn's disease, [and] other GI diseases that are related to immunological functions," Gesek says.
He adds that this understanding may lead to therapies that would either prevent the synthesis of the cytokine or keep it from interfering with sodium transport.
Related research by radiation oncologist Edward Abraham, M.D., has shown that CFTR and DF508-CFTR are expressed in the red blood cell membrane. He is now trying to identify drugs that will promote ciliary action and suppress sodium absorption in CF airways.
Bacterial invaders
Several DMS microbiologists are interested in the bacteria that infect CF airways. George O'Toole, Ph.D., is focusing on Pseudomonas aeruginosa, while Ambrose Cheung, M.D., is looking at Staphylococcus aureus.
Pseudomonas bacteria are everywhere and are usually harmless, except to people with CF. "There's something about a CF airway that [makes it] very susceptible to infection by Pseudomonas," says Stanton. "Once the Pseudomonas gets a foothold in the CF lung, it's almost impossible to eradicate it."
As for Staphylococcus, it's usually the first bacterium to invade and colonize the lungs of CF patients, often in infancy, setting the stage for later colonization by Pseudomonas. Cheung is tackling the staph problem with the help of two graduate students—self-described "geeks" Niles Donegan and Todd Jarry.
"We found that CF cells more readily take up bacteria than the normal cell line," says Cheung. "Maybe these bacteria hide inside epithelial cells . . . and start slowly multiplying. By hiding inside a cell, they are doing a couple of tricks. Number one, the antibody cannot get inside. Number two, neutrophils coming into the area are not going to recognize them, because they are inside the cell.
Donegan is trying to figure out which staph genes are activated when the bacteria burrow inside the epithelial cells. He's using a fluorescent green dye that acts like a molecular beacon—when a gene is activated, the beacon turns on. "We're putting these random pieces of DNA in front of this green dye protein," Donegan explains with enthusiasm. He doesn't seem to mind that there might be as many as 2,000 or 3,000 DNA sequences to check out. "We'll take each individual gene and proceed to disrupt it or turn it off in the cell and see if there's any obvious effect when we look at cells—their morphologies, the colony changes, something like that. If it can't invade the cell anymore, we'll know that we've got an interesting piece of DNA. That's kind of like the Holy Grail of what we're looking for right now."
"The long term plan," puts in Cheung, "is to find some kind of collaboration with industry to try to design molecules which mimic or disrupt this kind of gene."
Jarry, on the other hand, is looking at how the staph bacteria are enclosed in protective, bubblelike vesicles when they invade a cell. "It seems like staph is evading the whole [immune] process somehow," says Jarry. "Maybe a lack of protein on the [vesicle] membrane is stopping it from fusing to the lysosome that would kill it." Cheung's lab was the first to make this observation, and he expected publication of the finding in September.
O'Toole's lab is studying biofilm formation by Pseudomonas. "Biofilms are communities of bacteria that are attached to surfaces," says O'Toole. "They have a very unique architecture and structure, where you have big clumps of bacteria . . . separated by channels that allow the flow of nutrients. Some people have actually compared it to a primitive tissue with a circulatory system."
During the past two decades, scientists have become aware that bacteria grow predominantly in biofilm form in their natural world—in industrial, environmental, and medical settings. However, most of what is known about bacteria is based on how they grow in a free-swimming state. "No one has really tried to understand the molecular basis for the transition of bacteria from this planktonic, or free-swimming, mode of life to a mode of life where they're attached to a surface," O'Toole explains. Bacteria can be 500 to 1,000 times more resistant to antibiotics in a biofilm than in a free-swimming form, he says.
O'Toole hopes to learn why. "The thing that I find most appealing is that we're looking at a biological system that not much is known about," he says. "We have the potential to identify important genes that play a role in biofilm formation, and those genes are all targets for new therapeutic drugs. I think there's the potential there, whether it's [for] CF patients or helping cancer patients who are immunocompromised and getting colonized by bacteria biofilms." It's thought that 60 to 80 percent of hospital-acquired infections are derived from bacterial biofilms, he adds.
|
|
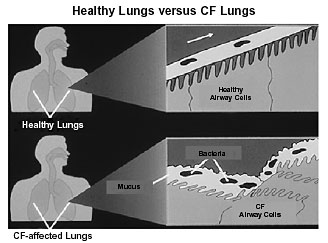
In both healthy and CF-affected lungs, the airway cells have tiny, hairlike projections called cilia. In healthy lungs, the fluid lining the airway traps potentially harmful substances, and the cilia beat in a coordinated action to sweep the foreign substances out. But in CF lungs, the airway fluid is mostly a mucus so thick and sticky that the cilia can hardly move. Bacteria stay trapped and can eventually cause infections. |
Other DMS researchers are investigating bacteria, too: toxicologist Aaron Barchowsky, Ph.D., is looking at the effects of environmental particles in the immune response in CF cells, and microbiologist Ronald Taylor, Ph.D., is characterizing the molecular mechanisms that mediate bacteria colonization and trying to determine how the environment within a human host may stimulate bacteria to express specific genes.
Outcomes orientation
"There are about a dozen or so research development programs in the country funded by the CF Foundation," Stanton says. "But Dartmouth was the first place to apply outcomes research to CF. We're now playing a national role in helping other programs develop an outcomes component. That's one of our major strengths."
Gerald O'Connor, Ph.D., associate director of DMS's Center for the Evaluative Clinical Sciences, heads up Dartmouth's CF outcomes group. Its key research tool is the Northern New England Cystic Fibrosis Consortium, a national model for outcomes research in the CF field. In addition to Dartmouth, the consortium includes Central Maine Medical Center in Lewiston, Eastern Maine Medical Center in Bangor, Maine Medical Center in Portland, and Fletcher Allen Health Care in Burlington, Vt. "It's a regional, voluntary, multidisciplinary group of clinicians and health-care professionals who seek to continuously improve the quality, safety, effectiveness, and costs of care for people with cystic fi- brosis," explains O'Connor.
Interdisciplinary work-groups within the consortium are exploring issues like laboratory procedures, aggressive treatment strategies, risk-prediction methods, nutrition, the effect of weight decline on long- and short-term outcomes, and the collection and use of quality-of-life data.
"On laboratory procedures, what we saw was wild variability in the rates of specific kinds of infections," says O'Connor. But no one was sure if these were real differences or just differences in lab procedures. The microbiology work group has "developed laboratory protocols across the system" in an attempt to answer that question.
The pulmonary group is assessing the effectiveness of aggressive treatment strategies. For example, the use of the mucus-thinning drug Pulmozyme varies dramatically throughout our country, yet "there's data from Denmark showing that more aggressive therapy is a better idea," O'Connor says. "There are probably some lessons to be learned."
Psychologist Mark Detzer, Ph.D., is developing standardized questionnaires for both patients and their parents to measure quality of life. "Teenagers and parents think very differently and observe very different things about . . . health care, self-care, disease management, [and] symptoms," he says. Having a structured way to get at this information will be helpful to clinicians in assessing how patients are doing emotionally as well as physically.
"Incredibly exciting"
What all these researchers and clinicians have in common is their passion for their work and their desire to help those suffering from CF.
"It's incredibly exciting to know that everything you do in the basic research lab today can be turned into . . . a therapeutic approach," says George O'Toole. "There are lots of times when you can take basic research and apply it right away—it really doesn't get any better than that."
For his part, Josh Hamilton says he's driven by "the challenge of finding something new and solving a problem. I like serendipity. Nature is always more clever and interesting than scientists."
And grad student Rangan Maitra, who may see yet another leap in the understanding and treatment of CF during his still-nascent career, likes "tangible projects which answer basic questions about how life functions. That's the kind of science that excites me," he says, "versus looking at a black hole or something that may not have any immediate impact on our lives. What I'd like to know is why are we the way we are."
|
|
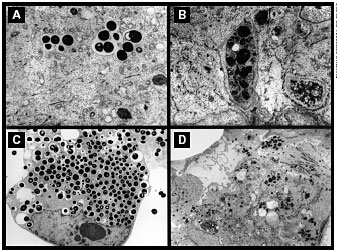
When staph bacteria invade the lungs
of a CF patient, they are enclosed in
protective vesicles—giving them the
appearance of black-yolked fried eggs
in this microscopic image. DMS
researchers are trying to determine
what aspect of the vesicle membrane
allows the bacteria to evade the
normal cell defense process. Frame A
shows live bacteria in a CF cell at 4
hours and Frame C shows how they've
multiplied after 24 hours. But the
normal defense process works if the
bacteria are heat-killed before being
placed in the CF cell, as in Frame B at
4 hours and Frame D at 24 hours. |
Laura Carter is the associate editor of Dartmouth Medicine magazine.
Back to Fall 2000 Dartmouth Medicine